The Shell
Adapted from the lesson by Tracy Teal. Original contributors: Paul Wilson, Milad Fatenejad, Sasha Wood and Radhika Khetani for Software Carpentry (http://software-carpentry.org/)
Learning Objectives
- What is the shell?
- How do you access it?
- How do you use it?
- Getting around the Unix file system
- looking at files
- manipulating files
- automating tasks
- What is it good for?
- Where are resources where I can learn more? (because the shell is awesome)
What is the shell?
The shell is a program that presents a command line interface which allows you to control your computer using commands entered with a keyboard instead of controlling graphical user interfaces (GUIs) with a mouse/keyboard combination.
There are many reasons to learn about the shell.
- For most bioinformatics tools, you have to use the shell. There is no graphical interface. If you want to work in metagenomics or genomics you’re going to need to use the shell.
- The shell gives you power. The command line gives you the power to do your work more efficiently and more quickly. When you need to do things tens to hundreds of times, knowing how to use the shell is transformative.
- To use remote computers or cloud computing, you need to use the shell.
Unix is user-friendly. It’s just very selective about who its friends are.
Today we’re going to go through how to access Unix/Linux and some of the basic shell commands.
Information on the shell
shell cheat sheets:
* http://fosswire.com/post/2007/08/unixlinux-command-cheat-sheet/
* https://github.com/swcarpentry/boot-camps/blob/master/shell/shell_cheatsheet.md
Explain shell - a web site where you can see what the different components of
a shell command are doing.
* http://explainshell.com
* http://www.commandlinefu.com
How to access the shell
We will be using the shell that’s available on Amazon EC2 (linux)
to launch EC2 instance:
Mac
On Mac, you need to start up Terminal
Applications -> Utilities -> Terminal
Go ahead and drag the Terminal application to your Dock for easy access.
Windows
For Windows, we’re going to be using MobaXterm.
Open up the program.
Linux
You need to start up Terminal
Launch EC2
ssh dcuser@LINK_FROM_ETHERPAD
Your computer will be unable to verify the authenticity of the host… type yes to continue connecting
Then enter the password for the remote instance: ‘data4Carp’ (Note: cursor will not move)
Starting with the shell
We will spend most of our time learning about the basics of the shell by manipulating some experimental data.
Now we’re going to download the data for the tutorial. For this you’ll need internet access, because you’re going to get it off the web.
We’re going to be working with data on our remote server.
After loggin on, let’s check out the example data.
Let’s go into the sample data directory
$ cd dc_sample_data
‘cd’ stands for ‘change directory’
Let’s see what is in here. Type ls
You will see:
sra_metadata untrimmed_fastq
ls stands for ‘list’ and it lists the contents of a directory.
There are two items listed. What are they? We can use a command line argumant with ‘ls’ to get more information.
$ ls -F
sra_metadata/ untrimmed_fastq/
Anything with a / after it is a directory.
Things with a * after them are programs.
It there are nodecorations, it’s a file.
You can also use the command
$ ls -l
drwxr-x--- 2 dcuser dcuser 4096 Jul 30 11:37 sra_metadata
drwxr-xr-x 2 dcuser dcuser 4096 Jul 30 11:38 untrimmed_fastq
to see whether items in a directory are files or directories. ls -l gives a lot more
information too.
Let’s go into the untrimmed_fastq directory and see what is in there.
$ cd untrimmed_fastq
$ ls -F
SRR097977.fastq SRR098026.fastq
There are two items in this directory with no trailing slash, so they are files.
Arguments
Most programs take additional arguments that control their exact
behavior. For example, -F and -l are arguments to ls. The ls
program, like many programs, take a lot of arguments. Another useful one is ‘-a’,
which show everything, including hidden files. How do we
know what the options are to particular commands?
Most commonly used shell programs have a manual. You can access the
manual using the man program. Try entering:
$ man ls
This will open the manual page for ls. Use the space key to go
forward and b to go backwards. When you are done reading, just hit q
to quit.
Programs that are run from the shell can get extremely complicated. To
see an example, open up the manual page for the find program.
No one can possibly learn all of
these arguments, of course. So you will probably find yourself
referring back to the manual page frequently.
The Unix directory file structure (a.k.a. where am I?)
As you’ve already just seen, you can move around in different directories or folders at the command line. Why would you want to do this, rather than just navigating around the normal way.
When you’re working with bioinformatics programs, you’re working with your data and it’s key to be able to have that data in the right place and make sure the program has access to the data. Many of the problems people run in to with command line bioinformatics programs is not having the data in the place the program expects it to be.
Moving around the file system
Let’s practice moving around a bit.
We’re going to work in that dc_sample_data directory.
First we did something like go to the folder of our username. Then we opened ‘dc_sample_data’ then ‘data’
Let’s draw out how that went.
Now let’s draw some of the other files and folders we could have clicked on.
This is called a hierarchical file system structure, like an upside down tree with root (/) at the base that looks like this.
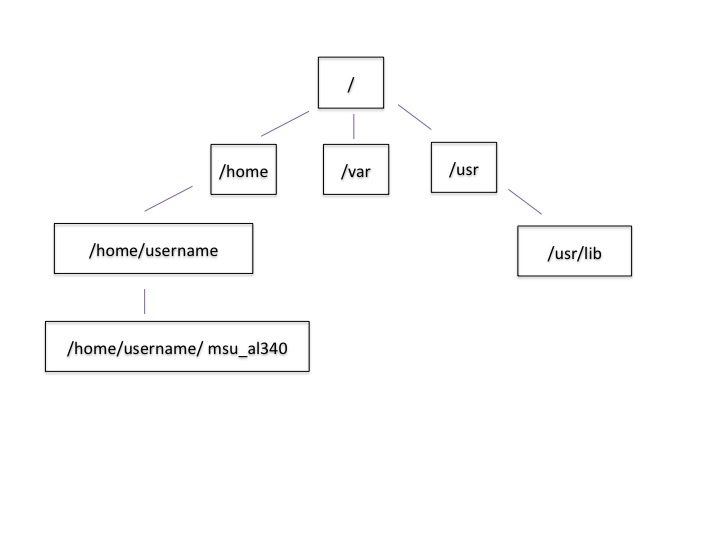
That (/) at the base is often also called the ‘top’ level.
When you are working at your computer or log in to a remote computer, you are on one of the branches of that tree, your home directory (/home/dcuser)
Now let’s go do that same navigation at the command line.
Type
$ cd
This puts you in your home directory. This folder here.
Now using cd and ls, go in to the ‘dc_sample_data’ directory and list its contents.
Let’s also check to see where we are. Sometimes when we’re wandering around in the file system, it’s easy to lose track of where we are and get lost.
If you want to know what directory you’re currently in, type
$ pwd
This stands for ‘print working directory’. The directory you’re currently working in.
What if we want to move back up and out of the ‘data’ directory? Can we just
type cd dc_sample_data? Try it and see what happens.
To go ‘back up a level’ we need to use ..
Type
$ cd ..
Now do ls and pwd. See now that we went back up in to the ‘dc_sample_data’
directory. .. means go back up a level.
Exercise
Now we’re going to try a hunt. Find a hidden directory in dc_sample_data list its contents and file the text file in there. What is the name of the file?
Hint: hidden files and folders in unix start with ‘.’, for example .my_hidden_directory
Examining the contents of other directories
By default, the ls commands lists the contents of the working
directory (i.e. the directory you are in). You can always find the
directory you are in using the pwd command. However, you can also
give ls the names of other directories to view. Navigate to the
home directory if you are not already there.
Type:
$ cd
Then enter the command:
$ ls dc_sample_data
This will list the contents of the dc_sample_data directory without
you having to navigate there.
The cd command works in a similar way. Try entering:
$ cd
$ cd dc_sample_data/untrimmed_fastq
and you will jump directly to untrimmed_fastq without having to go through
the intermediate directory.
Exercise
List the ‘SRR097977.fastq’ file from your home directory without changing directories
Shortcut: Tab Completion
Navigate to the home directory. Typing out directory names can waste a
lot of time. When you start typing out the name of a directory, then
hit the tab key, the shell will try to fill in the rest of the
directory name. For example, type cd to get back to your home directy, then enter:
$ cd dc_<tab>
The shell will fill in the rest of the directory name for
dc_sample_data. Now go to dc_sample_data/untrimmed_fastq
$ ls SR<tab><tab>
When you hit the first tab, nothing happens. The reason is that there
are multiple directories in the home directory which start with
SR. Thus, the shell does not know which one to fill in. When you hit
tab again, the shell will list the possible choices.
Tab completion can also fill in the names of programs. For example,
enter e<tab><tab>. You will see the name of every program that
starts with an e. One of those is echo. If you enter ec<tab> you
will see that tab completion works.
Full vs. Relative Paths
The cd command takes an argument which is the directory
name. Directories can be specified using either a relative path or a
full path. The directories on the computer are arranged into a
hierarchy. The full path tells you where a directory is in that
hierarchy. Navigate to the home directory. Now, enter the pwd
command and you should see:
/home/dcuser
which is the full name of your home directory. This tells you that you
are in a directory called dcuser, which sits inside a directory called
home which sits inside the very top directory in the hierarchy. The
very top of the hierarchy is a directory called / which is usually
referred to as the root directory. So, to summarize: dcuser is a
directory in home which is a directory in /.
Now enter the following command:
$ cd /home/dcuser/dc_sample_data/.hidden
This jumps to .hidden. Now go back to the home directory (cd). We saw
earlier that the command:
$ cd dc_sample_data/.hidden
had the same effect - it took us to the hidden directory. But,
instead of specifying the full path
(/home/dcuser/dc_sample_data/data), we specified a relative path. In
other words, we specified the path relative to our current
directory. A full path always starts with a /. A relative path does
not.
A relative path is like getting directions from someone on the street. They tell you to “go right at the Stop sign, and then turn left on Main Street”. That works great if you’re standing there together, but not so well if you’re trying to tell someone how to get there from another country. A full path is like GPS coordinates. It tells you exactly where something is no matter where you are right now.
You can usually use either a full path or a relative path depending on what is most convenient. If we are in the home directory, it is more convenient to just enter the relative path since it involves less typing.
Over time, it will become easier for you to keep a mental note of the structure of the directories that you are using and how to quickly navigate amongst them.
Exercise
Now, list the contents of the /bin directory. Do you see anything
familiar in there?
How can you tell these are programs rather than plain files?
Saving time with shortcuts, wild cards, and tab completion
Shortcuts
There are some shortcuts which you should know about. Dealing with the
home directory is very common. So, in the shell the tilde character,
“”~””, is a shortcut for your home directory. Navigate to the dc_sample_data
directory:
$ cd
$ cd dc_sample_data
Then enter the command:
$ ls ~
This prints the contents of your home directory, without you having to
type the full path. The shortcut .. always refers to the directory
above your current directory. Thus:
$ ls ..
prints the contents of the /home/dcuser/dc_sample_data. You can chain
these together, so:
$ ls ../../
prints the contents of /home/dcuser which is your home
directory. Finally, the special directory . always refers to your
current directory. So, ls, ls ., and ls ././././. all do the
same thing, they print the contents of the current directory. This may
seem like a useless shortcut right now, but we’ll see when it is
needed in a little while.
To summarize, while you are in the shell directory, the commands
ls ~, ls ~/., ls ../../, and ls /home/dcuser all do exactly the
same thing. These shortcuts are not necessary, they are provided for
your convenience.
Our data set: FASTQ files
We did an experiment and want to look at sequencing results. We want to be able to look at these files and do some things with them.
Wild cards
Navigate to the ~/dc_sample_data/data/untrimmed_fastq directory. This
directory contains our FASTQ files.
The * character is a shortcut for “everything”. Thus, if
you enter ls *, you will see all of the contents of a given
directory. Now try this command:
$ ls *fastq
This lists every file that ends with a fastq.
This command:
$ ls /usr/bin/*.sh
Lists every file in /usr/bin that ends in the characters .sh.
This command:
$ ls *977.fastq
lists only the file that ends with ‘977.fastq’
So how does this actually work? Well…when the shell (bash) sees a
word that contains the * character, it automatically looks for filenames
that match the given pattern.
We can use the command ‘echo’ to see wilcards are they are intepreted by the shell.
$echo *.fastq
SRR097977.fastq SRR098026.fastq
The ‘*’ is expanded to include any file that ends with ‘.fastq’
Exercise
Do each of the following using a single ls command without
navigating to a different directory.
- List all of the files in
/binthat start with the letter ‘c - List all of the files in
/binthat contain the letter ‘a’ - List all of the files in
/binthat end with the letter ‘o’
BONUS: List all of the files in ‘/bin’ that contain the letter ‘a’ or ‘c’
Command History
You can easily access previous commands. Hit the up arrow. Hit it again. You can step backwards through your command history. The down arrow takes your forwards in the command history.
^-C will cancel the command you are writing, and give you a fresh prompt.
^-R will do a reverse-search through your command history. This is very useful.
You can also review your recent commands with the history command. Just enter:
$ history
to see a numbered list of recent commands, including this just issues
history command. You can reuse one of these commands directly by
referring to the number of that command.
If your history looked like this:
259 ls *
260 ls /usr/bin/*.sh
261 ls *R1*fastq
then you could repeat command #260 by simply entering:
!260
(that’s an exclamation mark). You will be glad you learned this when you try to re-run very complicated commands.
Exercise
- Find the line number in your history for the last exercise (listing
files in
/bin) and reissue that command.
Examining Files
We now know how to switch directories, run programs, and look at the contents of directories, but how do we look at the contents of files?
The easiest way to examine a file is to just print out all of the
contents using the program cat. Enter the following command:
$ cat SRR098026.fastq
This prints out the all the contents of the the SRR098026.fastq to the screen.
Exercises
-
Print out the contents of the
~/dc_sample_data/untrimmed_fastq/SRR097977.fastqfile. What does this file contain? -
From your home directory, without changing directories, use one short command to print the contents of all of the files in the
/home/dcuser/dc_sample_data/untrimmed_fastqdirectory.
$ cd ~/dc_sample_data/untrimmed_fastq
cat is a terrific program, but when the file is really big, it can
be annoying to use. The program, less, is useful for this
case. Enter the following command:
$ less SRR098026.fastq
less opens the file, and lets you navigate through it. The commands
are identical to the man program.
Some commands in less
| key | action |
|---|---|
| “space” | to go forward |
| “b” | to go backwarsd |
| “g” | to go to the beginning |
| “G” | to go to the end |
| “q” | to quit |
less also gives you a way of searching through files. Just hit the
“/” key to begin a search. Enter the name of the word you would like
to search for and hit enter. It will jump to the next location where
that word is found. Try searching the dictionary.txt file for the
word “cat”. If you hit “/” then “enter”, less will just repeat
the previous search. less searches from the current location and
works its way forward. If you are at the end of the file and search
for the word “cat”, less will not find it. You need to go to the
beginning of the file and search.
For instance, let’s search for the sequence GTGCGGGCAATTAACAGGGGTTCAC in our file.
You can see that we go right to that sequence and can see
what it looks like.
Remember, the man program actually uses less internally and
therefore uses the same commands, so you can search documentation
using “/” as well!
There’s another way that we can look at files, and in this case, just look at part of them. This can be particularly useful if we just want to see the beginning or end of the file, or see how it’s formatted.
The commands are head and tail and they just let you look at
the beginning and end of a file respectively.
$ head SRR098026.fastq
$ tail SRR098026.fastq
The -n option to either of these commands can be used to print the
first or last n lines of a file. To print the first/last line of the
file use:
$ head -n 1 SRR098026.fastq
$ tail -n 1 SRR098026.fastq
Creating, moving, copying, and removing
Now we can move around in the file structure, look at files, search files, redirect. But what if we want to do normal things like copy files or move them around or get rid of them. Sure we could do most of these things without the command line, but what fun would that be?! Besides it’s often faster to do it at the command line, or you’ll be on a remote server like Amazon where you won’t have another option.
Our raw data in this case is fastq files. We don’t want to change the original files, so let’s make a copy to work with.
Lets copy the file using the cp command. The cp
command backs up the file. Navigate to the data directory and enter:
$ cp SRR098026.fastq SRR098026-copy.fastq
$ ls -F
SRR097977.fastq SRR098026-copy.fastq SRR098026.fastq
Now ‘SRR098026-copy.fastq’ has been created as a copy of ‘SRR098026.fastq’
Let’s make a backup directory where we can put this file.
The mkdir command is used to make a directory. Just enter mkdir
followed by a space, then the directory name.
$ mkdir backup
We can now move our backed up file in to this directory. We can
move files around using the command mv. Enter this command:
$ mv *-copy.fastq backup
$ ls -al backup
total 52
drwxrwxr-x 2 dcuser dcuser 4096 Jul 30 15:31 .
drwxr-xr-x 3 dcuser dcuser 4096 Jul 30 15:31 ..
-rw-r--r-- 1 dcuser dcuser 43421 Jul 30 15:28 SRR098026-copy.fastq
The mv command is also how you rename files. Since this file is so
important, let’s rename it:
$ cd backup
$ mv SRR098026-copy.fastq SRR098026-copy.fastq_DO_NOT_TOUCH!
$ ls
SRR098026-copy.fastq_DO_NOT_TOUCH!
Finally, we decided this was silly and want to start over.
$ rm backup/SRR*
The rm file permanently removes the file. Be careful with this command. It doesn’t
just nicely put the files in the Trash. They’re really gone.
Exercise
Do the following:
- Create a backup of your fastq files
- Create a backup directory
- Copr your backup files there
By default, rm, will NOT delete directories. You can tell rm to
delete a directory using the -r option. Let’s delete that new directory
we just made. Enter the following command:
$ rm -r backup
Writing files
We’ve been able to do a lot of work with files that already exist, but what if we want to write our own files. Obviously, we’re not going to type in a FASTA file, but you’ll see as we go through other tutorials, there are a lot of reasons we’ll want to write a file, or edit an existing file.
To write in files, we’re going to use the program nano. We’re going to create
a file that contains the favorite grep command so you can remember it for later. We’ll name this file
‘awesome.sh’.
$ nano awesome.sh
Now you have something that looks like
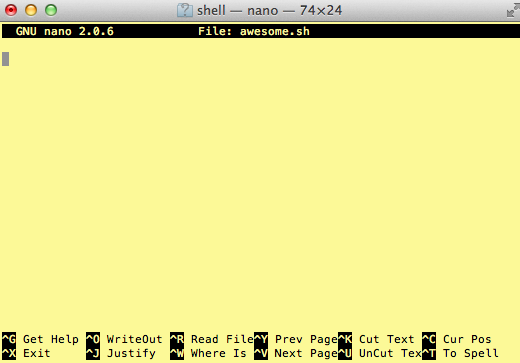
Type in your command, so it looks like
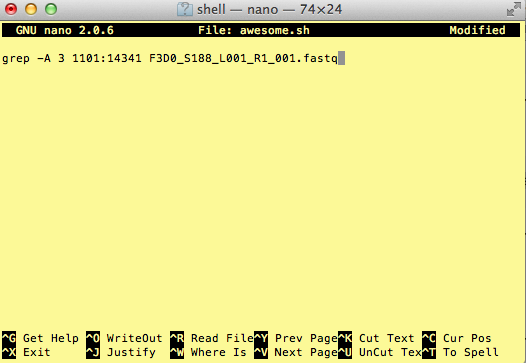
Now we want to save the file and exit. At the bottom of nano, you see the “^X Exit”. That
means that we use Ctrl-X to exit. Type Ctrl-X. It will ask if you want to save it. Type y for yes.
Then it asks if you want that file name. Hit ‘Enter’.
Now you’ve written a file. You can take a look at it with less or cat, or open it up again and edit it.
Exercise
Open ‘awesome.sh’ and add “echo AWESOME!” after the grep command and save the file.
We’re going to come back and use this file in just a bit.